Getting an AI to write a Clinical Study Report section is no longer the hard part. Getting a section that a regulator will actually accept is. The gap between "a draft exists" and "a reviewer signed off" is where medical-writing teams still spend their nights — running a section past the ICH E3 structure, checking that adverse events are ordered the way FDA expects, confirming the primary analysis is reported in full and not deferred, making sure a cross-reference to a table actually resolves.
That work has always happened after the draft, in a different tool, in a different person's head, days later. Asthra's newest capability moves it into the draft. It's called Regulatory Review, and it puts a US FDA / ICH reviewer's eye on every section, on demand, right where the writer is working — and it grounds every recommendation in two things AI usually hand-waves: curated regulatory guidance and real approved-product precedent pulled live from FDA's own database.
This post is a look under the hood at how it works, and why we built it the way we did.
A quick clarification up front, because the word "reviewer" is overloaded: Regulatory Review is not a chatbot persona role-playing an inspector. It's a deterministic audit pass — curated guidance in, real FDA precedent alongside, structured findings out, planted in the document. It's a different thing from the configurable reviewer-persona scorecards on our roadmap, and it stands on its own.
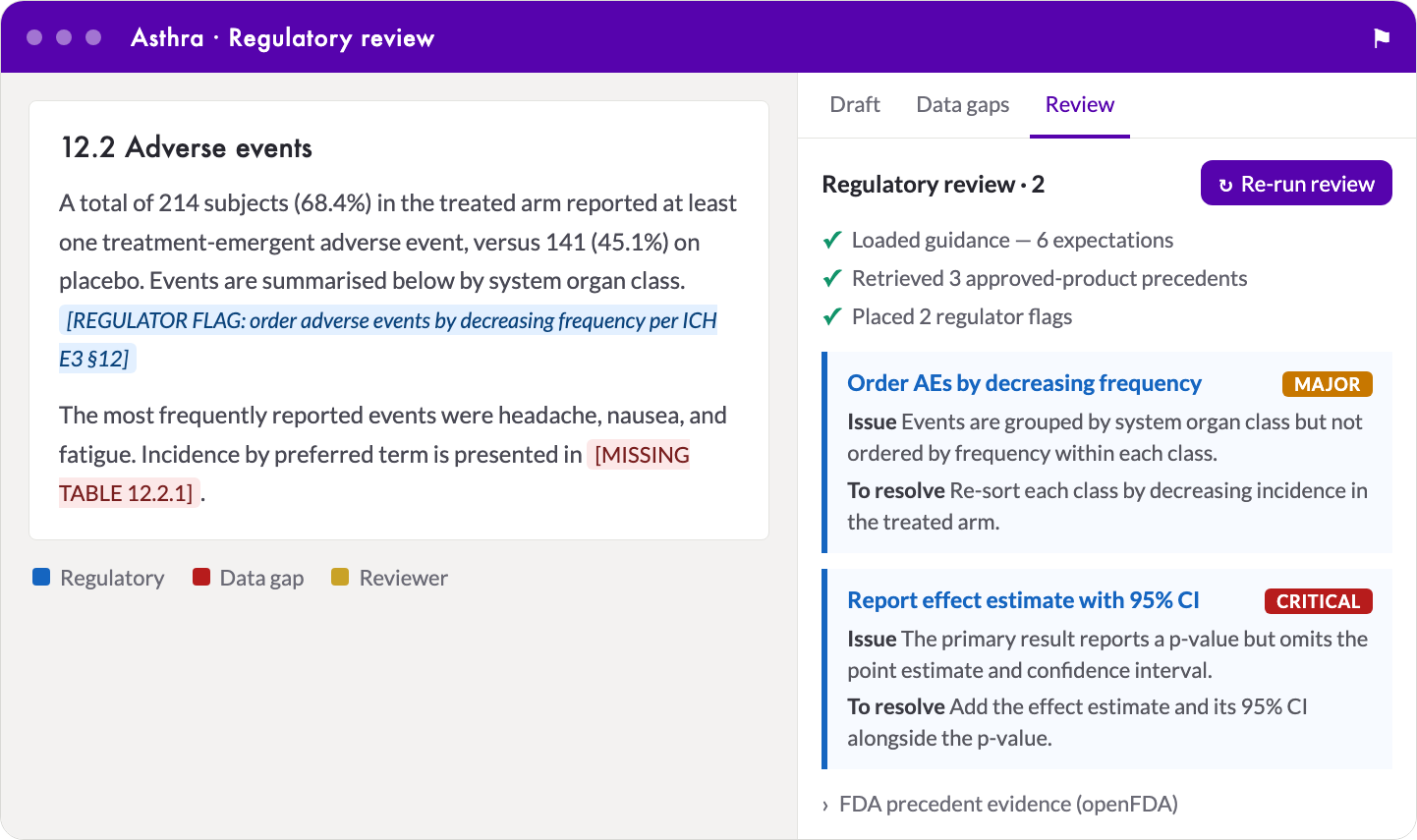 Regulatory Review inside the draft: inline
Regulatory Review inside the draft: inline [REGULATOR FLAG] (blue) and data-gap (red) markers sit in the section on the left; the audit panel on the right shows the three streamed steps, the severity-ranked findings, and the openFDA precedent behind each.
The problem: "sounds regulatory" is not the same as "is compliant"
A frontier LLM will happily produce a CSR section that reads like a CSR section. It has the right cadence, the right vocabulary, the right headings. That's exactly the trap. Fluency is cheap; compliance is specific. A regulator doesn't grade prose — they check a section against a concrete set of expectations:
- Is the primary efficacy analysis reported in full, or quietly deferred to a later module?
- Are adverse events presented by decreasing frequency, per ICH E3 §12?
- Is the effect estimate reported with its 95% confidence interval?
- Does every reference to a table or figure resolve to an artefact that actually exists in the package?
- Are subgroup findings labelled as pre-specified or post-hoc?
None of that is a matter of style. And none of it is something you can trust a general-purpose model to check by "reading carefully," because the model has no anchor for what this section, in this document type, is supposed to contain. It needs a rubric, and it needs precedent. Regulatory Review gives it both.
One click, three transparent steps
From the writer's side, the feature is deliberately unglamorous. In the section inspector — the panel that sits next to the live Word document — there's a Review tab with a single button: Run regulatory review. Click it on any drafted section and the audit runs against just that section.
What happens next is streamed back in three visible steps, so the writer is never staring at a spinner wondering what the AI is doing:
| Step | What the writer sees | What's happening underneath |
|---|---|---|
| 1 | "Loaded guidance for Clinical Efficacy Results (6 expectations)" | The curated FDA/ICH requirement checklist for this exact section is loaded |
| 2 | "Retrieved 3 approved-product precedent(s)" | openFDA is queried for real approved-label precedent |
| 3 | "2 regulatory flags" | The audit LLM has compared the draft to the expectations and placed its findings |
Each step is a real server-sent event (audit_guidelines_loaded, audit_precedents_fetched, audit_complete), not a decorative animation. Transparency is a feature here, not a nicety — in a regulated workflow, a writer needs to know what the machine checked before they trust whether it passed.
Step one: curated guidance, kept in sync with the drafting instructions
The audit doesn't invent its expectations. Each document type ships with a curated regulatory-guidance file — for CSR, that's an ICH E3–anchored document maintained alongside the templates. The loader slices it by section using the same heading the drafting instructions use, so the expectations an auditor checks are always in lockstep with the guidance the writer was given. There's no drift between "what we asked the draft to do" and "what we grade it against."
A section's guidance is two things stacked together: human-readable ICH E3 prose about what the section should cover, and — optionally — a structured requirement checklist that makes the expectations machine-checkable:
requirements:
- id: "pop-ae-frequency"
text: "Report adverse events ordered by decreasing frequency"
severity: "major"
- id: "efficacy-effect-estimate"
text: "Report the primary effect estimate with its 95% confidence interval"
severity: "critical"
openfda:
label_sections: ["adverse_reactions", "clinical_studies"]
search_terms: ["{indication}"]
Each requirement carries its own severity — critical, major, or minor — so the finding a writer sees inherits the regulatory weight of the expectation, not a weight the model guessed at. And when a section has no bespoke guidance yet, the auditor falls back to a set of generic ICH E3 expectations (completeness, results by treatment group with precision, neutral language, resolvable cross-references, documented data gaps) rather than silently doing nothing.
Step two: grounding in real FDA precedent, not the model's memory
This is the part that separates Regulatory Review from "ask an LLM if this looks compliant."
Before the audit runs, the system figures out what drug and indication the document is about — not by asking the writer to type it, but by querying the document's own source library ("What is the investigational compound?", "What is the indication?") and extracting a structured {drug_name, indication} from the answer. That inference is cached per run, so auditing a second section doesn't re-do the work.
With drug and indication in hand, it queries openFDA — FDA's public API — at the drug Structured Product Labeling endpoint (/drug/label.json), building a Lucene query against the brand name and indication. It pulls up to five approved-product label records and extracts the sections that matter for the audit — indications_and_usage, clinical_studies, adverse_reactions — and hands them to the reviewer as precedent: the public proxy for "what a filed, approved version of this section looks like," since real CSRs aren't public.
The framing of that precedent is the important design decision. The prompt is explicit:
IMPORTANT: The openFDA content above is PRECEDENT from other approved products' public labeling. Use it only to judge whether this section presents the kinds of analyses/statements regulators expect to see. NEVER import another product's numbers, populations, or claims into your findings.
Precedent tells the reviewer what a filed, approved version of this section looks like — how efficacy is stated, how safety is framed, what a real label includes. It does not become a source of borrowed data. That guardrail is the difference between "grounded" and "contaminated." And the whole precedent step is fail-soft: if openFDA is unreachable, or the drug can't be inferred, the audit simply proceeds on curated guidance alone, with a note — it never blocks, and it never fabricates.
Step three: the reviewer that recommends but never rewrites
Now the audit LLM does its job. Its instructions are narrow and specific:
You are a US FDA regulatory reviewer auditing a single section of a Clinical Study Report (CSR) drafted per ICH E3. Your job is to judge whether the drafted section satisfies regulatory expectations and to recommend concrete drafting actions — NOT to rewrite the section.
For every expectation, it decides whether the draft addresses it (present), partially addresses it (partial), or omits it (absent) — and it reports only the absent and partial ones. Anything already handled well is left alone. The writer isn't flooded with confirmations of what they got right; they get a short list of what's still missing.
Each finding comes back as a self-contained mini-report — strict JSON, no prose — that a medical writer can act on without further translation:
| Field | What it holds |
|---|---|
requirement | A short, actionable title (becomes the inline flag label) |
issue | What's missing or inadequate in this draft, and why a regulator would care |
resolution | The concrete step to fix it — what to add, restructure, or report |
severity | critical | major | minor, inherited from the requirement |
status | present | partial | absent |
anchor | A verbatim phrase from the draft, so the flag can be placed exactly where it applies |
Crucially, the reviewer never edits the writer's words. It recommends. The separation between "an agent that reviews" and "an agent that writes" is intentional — a reviewer that silently rewrites is impossible to trust and impossible to audit. Asthra keeps the two roles distinct.
Where the findings actually land: inside the document
A review panel full of findings is useful. A review panel whose findings live in the document, at the exact spot they apply is a different class of tool.
Every actionable finding is turned into an inline marker in the live draft:
[REGULATOR FLAG: present adverse events by decreasing frequency per ICH E3 §12]
These markers are rendered with soft blue shading so a writer can scan a section and see every outstanding regulatory recommendation at a glance. Placement isn't approximate: the system uses the finding's anchor to locate the exact paragraph the requirement applies to — first by exact substring match, then by fuzzy token overlap (≥60%, skipping headings and table rows) — and drops the marker there. If a requirement is about content that's entirely missing (so there's nothing to anchor to), the marker goes into a clearly labelled "Regulatory review recommendations" block at the end of the section instead of guessing.
And the whole reconciliation is idempotent. Re-run the review after fixing three of five flags, and it strips every existing marker and re-inserts exactly the findings that still apply — no duplicates, no stale flags, no drift between what the panel says and what's in the document. In the review panel, each finding is a clickable card; clicking it jumps to and selects that marker in Word (with a three-tier fallback: the exact marker, then the anchor phrase, then the section heading), so "I see the issue" and "take me to it" are one motion.
A deliberate colour code: three kinds of "not done"
Regulatory Review ships alongside a companion surface — a new Data Gaps tab — and the relationship between them is a small but important piece of the design.
Not every "this section isn't finished" is the same kind of problem, and conflating them is how writers lose trust in a tool. Asthra keeps three distinct:
- Data gaps (red) — genuine source-data holes the drafting agent honestly flagged:
[MISSING INFO: …],[NOT FOUND — NOT IN SOURCE],[MISSING TABLE …]. The source material didn't contain what the section needs. The Data Gaps tab lists these per section and lets the writer click straight to each one in the document. - Regulatory recommendations (blue) — the
[REGULATOR FLAG: …]markers from this audit: the source data may be complete, but the presentation doesn't yet meet a regulatory expectation. Notably, these do not lower a section's completeness score — a recommendation to reorder AEs isn't a hole in the data. - Reviewer / honesty flags (yellow) — places the drafting agent itself surfaced for human attention.
Three colours, three meanings, three workflows. A missing table is a retrieval problem. A mis-ordered AE list is a drafting problem. Collapsing them into one undifferentiated "issues" list would make both harder to fix. Separating them is what lets a writer triage in seconds.
Built for a regulated workflow: idempotent, fail-soft, persistent
A few properties matter more in this domain than they would almost anywhere else, and they're baked in rather than bolted on:
- Fail-soft at every seam. openFDA down? Audit on guidance alone. Drug can't be inferred? Skip precedent, keep going. No curated guidance for this section? Use generic ICH E3 expectations. The model returns malformed JSON? Report a clean
llm_error, leave the draft untouched. The audit degrades; it never corrupts. - Idempotent by construction. Running the review twice never doubles a flag or leaves a resolved one behind. State converges to "exactly the findings that currently apply."
- Persistent across sessions. Findings, the FDA precedent evidence, the annotated draft, and a timestamp are stored with the run. Close the document and reopen it a week later and the flags are still there, re-loaded without re-spending a single token on a fresh audit. The section's spine even carries a small badge — the flag count and when it was last audited — so the state of review is visible before you open the tab.
- Recommends, never rewrites. The reviewer's output is advice with an anchor, not a silent edit. The writer stays in control of the words.
Why this is the shape of trustworthy regulatory AI
There's a pattern here that runs through everything Asthra builds. The hard problem in regulatory writing isn't generating text — it's making text defensible. Regulatory Review is defensible because every judgment it makes is traceable to something concrete: a curated ICH E3 expectation with a known severity, and real precedent from a real approved label that anyone can pull up on openFDA. It doesn't ask a writer to trust the model's taste. It shows its work, in three visible steps, and it plants its conclusions in the document where they can be checked, acted on, and re-checked.
An AI that drafts is a productivity tool. An AI that drafts and hands the draft to a grounded FDA reviewer before it ever leaves your desk — that's the beginning of a submission you can stand behind.